© imago stock/StockTrek Images
Charité-Expertin für seltene Krankheiten: Lena Oevermann hilft Menschen aus aller Welt
Die Sichelzellkrankheit ist in Europa selten, in anderen Ländern hingegen noch verbreitet. Oberärztin Lena Oevermann behandelt junge Patienten solchen genetisch bedingten Erkrankungen.
Seit fünf Jahren ist der junge Mann nun in Berlin. Mit 16 Jahren hat er sich aus Gambia aufgemacht, ist über Libyen und Italien nach Berlin gekommen, wo sein älterer Bruder schon lebte. Eine beschwerliche Reise, auch wegen seiner gesundheitlichen Probleme: Immer wieder schlimme Schmerzen, Herzschwäche, geschädigte Knochen. Er habe schon gewusst, was mit ihm los sei, sagte der Jugendliche in der Ambulanz des Zentrums für Hämoglobinerkrankungen der Charité Universitätsmedizin, wo er gezielt Hilfe suchte.
„Er kommt aus einem Land, in dem die Sichelzellkrankheit häufig ist, aber oft die Mittel fehlen, um sie zu behandeln“, sagt Lena Oevermann, Oberärztin an der Klinik für Hämatologie und Onkologie im Kindes- und Jugendalter und Leiterin des dort angesiedelten Zentrums. Ihre Arbeitsgruppe arbeitet mit mehreren Kliniken in Nairobi zusammen. Die Menschen dort leben seit jeher mit der Erfahrung, dass Betroffene jung an der Krankheit sterben.
350.000 Kinder kommen in jedem Jahr vor allem in Subsahara-Afrika, aber auch in einigen Ländern Asiens und in Südamerika mit der Veranlagung für diese Veränderung der roten Blutkörperchen zur Welt. In Europa ist die Sichelzellkrankheit eine seltene Erkrankung, durch die großen Migrationsbewegungen gewinnt sie aber auch bei uns an Bedeutung und Aufmerksamkeit.
Wir können bei der Sichelzellkrankheit die Eltern schulen, schon bevor es ein medizinisches Problem gibt.
Lena Oevermann, Fachärztin für Kinder- und Jugendmedizin
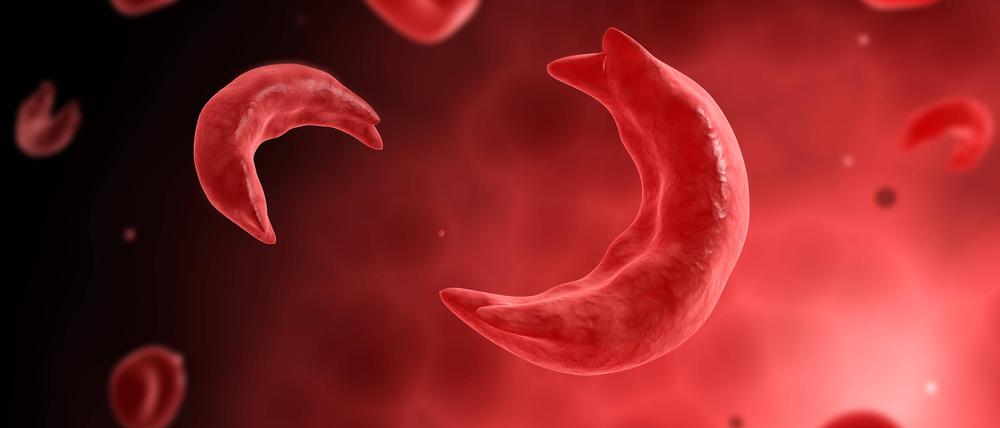
Bei der Krankheit, für die beim Kind die Anlage beider Eltern zusammenkommen muss, führt der Austausch einer einzigen Aminosäure dazu, dass sich „falscher“ roter Blutfarbstoff bildet. Das Hämoglobin verklebt, bildet Fasern, die die roten Blutkörperchen schädigen: Sie bekommen statt der gesunden runden die sichelförmige Form, die der Krankheit den Namen gab. Diese roten Blutkörperchen zerfallen schneller als gesunde, sodass es zur Blutarmut kommt, zur chronischen Anämie. Weil die Sauerstoffversorgung schlecht ist, leiden viele Organe. Zugleich verfangen sich die gebildeten Sichelzellen schneller in der Blutbahn, was Gefäßverschlüsse bis hin zu Schlaganfällen zur Folge haben kann.

© Charité Berlin
Seit zwei Jahren wird hierzulande beim Neugeborenen-Screening im Blut auch nach der Sichelzellkrankheit gesucht. Das ist gut, weil man im Falle eines Falles schon früh etwas tun kann. „Wir können die Eltern schulen, schon bevor es ein medizinisches Problem gibt“, sagt Oevermann. „Sie müssen unbedingt wissen, wann sie zum Arzt gehen und wann vielleicht sogar einen Krankenwagen rufen müssen.“
Stammzelltransplantation kann heilen
Zur Standardbehandlung gehört Hydroxycarbamid, ein Krebsmedikament, das hier aber niedrig dosiert eingesetzt wird und dafür sorgt, dass der Organismus mehr vom unbeeinträchtigten „fetalen“ Hämoglobin bildet, dessen Bedeutung sonst bei Kindern mit der Zeit abnimmt. Große Bedeutung haben zudem Bluttransfusionen, bis hin zum Austausch des gesamten Bluts.
In ihrer eigenen Forschung widmet sich Oevermann einer Behandlungsform, die die Chance auf Heilung bietet, der Stammzelltransplantation. Findet sich, im besten Fall, unter Geschwistern des Patient:innen, den allernächsten Verwandten, ein passender Spender dafür, und wird der Austausch der Blutzellen vor dem Schulalter vorgenommen, dann liegen die Erfolgschancen der Therapie bei 95 Prozent. Gefördert von Berlin Institute of Health (BIH) entwickelt Oevermanns Arbeitsgruppe derzeit mit Hilfe Künstlicher Intelligenz einen Algorithmus, um das „Matching“ zwischen Spendern, zu denen unter Umständen auch ein Elternteil oder ein „Fremdspender“ gehören können, zu optimieren. Grundstock ist eine eigene Biobank.

© privat
Zu den teuren Behandlungsansätzen gehören inzwischen auch viel versprechende Gentherapien, mit denen die genetische Besonderheit gezielt korrigiert werden kann. Oevermann findet es gut, dass Gentherapien und Stammzelltherapien inzwischen gegeneinander ins Rennen gehen können: Ein Ansporn zur Verbesserung für beide.
Auf Zypern gibt es Frühdiagnostik auf Thalassämie
Die Transplantation von Blutstammzellen wird auch bei einer anderen bei uns seltenen Erkrankung des Hämoglobins eingesetzt, bei der Thalassämie. Sie heißt nach dem griechischen Wort für „Meer“, da sie im südlichen Mittelmeerraum recht verbreitet ist. Bei der schweren Form dieser ebenfalls monogenetischen Erkrankung bilden schon Babys zu wenig Blutfarbstoff. Die Betroffenen haben weniger rote Blutkörperchen, die zudem kleiner sind, weniger Hämoglobin enthalten und den Körper nicht ausreichend mit Sauerstoff versorgen können. Die Kinder sind deshalb schnell erschöpft. Das Knochenmark versucht den Mangel zu kompensieren und wird überaktiv, es kommt zu schmerzhaften Veränderungen am Skelett.
Die Charité-Mediziner haben inzwischen bei der Stammzelltransplantation eine Kooperation mit Medizinern aus Zypern, wo die Thalassämie traditionell häufig ist. Fast jede siebte Person trägt die Anlage dafür, die meisten allerdings, ohne selbst erkrankt zu sein. Werden zwei Anlage-Träger ein Paar, dann könnten dessen Kinder jedoch erkranken.
Seit Jahren werden alle Männer und Frauen im griechischen Teil der Insel vor der Eheschließung verpflichtend getestet. Sie könnten sich, wenn beide Träger des veränderten Gens sind, im Wissen darum zum Beispiel für eine In-vitro-Fertilisation mit anschließender Diagnostik vor der Implantation einer befruchteten Eizelle entscheiden, um eine Erkrankung bei ihrem Kind auszuschließen. Einige Jahre lang seien kaum Kinder mit einer Thalassämie geboren worden, das habe sich inzwischen aber wieder geändert, berichtet Oevermann.
Zu dieser Veränderung könnten auch die verbesserten Behandlungsmöglichkeiten für Krankheiten der roten Blutkörperchen beigetragen haben. Von ihnen profitierte etwa der junge Mann aus Gambia, der mit 18 Jahren die Blutstammzellen seines Bruders transplantiert bekam. Er ist jetzt 21 Jahre alt, beginnt gerade eine Ausbildung und hat inzwischen schon zweimal vor Medizin-Studierenden in der Charité-Vorlesung über seinen eigenen Fall berichtet.
- showPaywall:
- false
- isSubscriber:
- false
- isPaid:
- showPaywallPiano:
- false